RDKit Interface¶
RDKit is a cheminformatics toolkit that provides extensive functionality for molecular manipulation, analysis, and property calculation.
RDKit molecules contain explicit bond information.
For more information about RDKit, visit https://www.rdkit.org/docs/index.html
[1]:
from IPython.display import display
from rdkit import Chem
import molify
RDKit → ASE: connectivity key.¶
When we convert from RDKit to ASE, all the bond information is preserved in the atoms.info['connectivity'] object and the initial charge state of the atoms object. The following example creates an acetic acid molecule to demonstrate this conversion.
[2]:
# Create acetic acid from SMILES
acetic_acid = Chem.MolFromSmiles("CC(=O)[O-]")
acetic_acid = Chem.AddHs(acetic_acid) # Add explicit hydrogens
# Display the molecule
print("Acetic acid molecule:")
display(acetic_acid)
Acetic acid molecule:
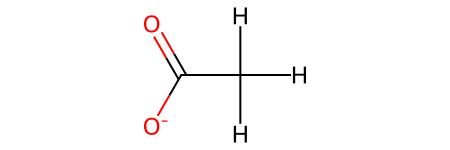
[3]:
# Convert to ASE
acetic_acid_atoms = molify.rdkit2ase(acetic_acid)
print(f"Number of atoms: {len(acetic_acid_atoms)}")
print(f"Chemical formula: {acetic_acid_atoms.get_chemical_formula()}")
print(f"\nAtoms object: {acetic_acid_atoms}")
print(f"Initial charge: {acetic_acid_atoms.get_initial_charges()}")
Number of atoms: 7
Chemical formula: C2H3O2
Atoms object: Atoms(symbols='C2O2H3', pbc=False, initial_charges=...)
Initial charge: [ 0. 0. 0. -1. 0. 0. 0.]
The connectivity Data Structure¶
The bond information is stored in atoms.info['connectivity'] as a list of tuples:
[(atom_idx_1, atom_idx_2, bond_order), ...]
Where:
atom_idx_1, atom_idx_2: 0-based integer atom indices
bond_order: Float representing bond type:
1.0= single bond2.0= double bond3.0= triple bond1.5= aromatic bondNone= unknown bond order
The following examines the connectivity:
[ ]:
# Display the connectivity information
connectivity = acetic_acid_atoms.info["connectivity"]
print("Connectivity structure:")
print(f"Total bonds: {len(connectivity)}\n")
for i, (atom1, atom2, bond_order) in enumerate(connectivity):
symbol1 = acetic_acid_atoms.get_chemical_symbols()[atom1]
symbol2 = acetic_acid_atoms.get_chemical_symbols()[atom2]
print(
f" Bond {i}: atom {atom1} ({symbol1}) - "
f"atom {atom2} ({symbol2}), order = {bond_order}"
)
[5]:
# The SMILES string is also stored for convenience
print(f"Original SMILES: {acetic_acid_atoms.info['smiles']}")
print(f"\nAll info keys: {list(acetic_acid_atoms.info.keys())}")
Original SMILES: [H]C([H])([H])C(=O)[O-]
All info keys: ['smiles', 'connectivity']
Key Insight¶
When converting from RDKit to ASE, all bond information is explicit:
✅ Bond connectivity is known
✅ Bond orders are known
✅ Formal charges are preserved
✅ SMILES string is stored
RDKit → NetworkX: Understanding Graph Structure¶
NetworkX provides a graph-based representation that’s useful for analyzing molecular topology, finding cycles, and understanding connectivity patterns.
The following converts the same acetic acid molecule to a NetworkX graph:
[6]:
import networkx as nx
# Convert to NetworkX graph
acetic_acid_graph = molify.rdkit2networkx(acetic_acid)
print(f"Number of nodes (atoms): {acetic_acid_graph.number_of_nodes()}")
print(f"Number of edges (bonds): {acetic_acid_graph.number_of_edges()}")
Number of nodes (atoms): 7
Number of edges (bonds): 6
Node Attributes: Atom Properties¶
Each node in the graph represents an atom with these attributes:
atomic_number: Element atomic number (e.g., 6 for carbon, 8 for oxygen)original_index: Index from the RDKit moleculecharge: Formal charge on the atom
The following examines a few nodes:
[7]:
for node_id in list(acetic_acid_graph.nodes):
attrs = acetic_acid_graph.nodes[node_id]
atomic_num = attrs["atomic_number"]
symbol = acetic_acid_graph.nodes[node_id]["atomic_number"]
print(f" Node {node_id}: {symbol} (Z={atomic_num}), charge={attrs['charge']}")
Node 0: 6 (Z=6), charge=0
Node 1: 6 (Z=6), charge=0
Node 2: 8 (Z=8), charge=0
Node 3: 8 (Z=8), charge=-1
Node 4: 1 (Z=1), charge=0
Node 5: 1 (Z=1), charge=0
Node 6: 1 (Z=1), charge=0
Edge Attributes: Bond Information¶
Each edge represents a bond with:
bond_order: The bond type (1.0, 2.0, 3.0, or 1.5 for aromatic)
When converting from RDKit, bond_order is always present.
[ ]:
# Display edge attributes (bonds)
for i, (node1, node2, attrs) in enumerate(list(acetic_acid_graph.edges(data=True))):
symbol1 = acetic_acid_graph.nodes[node1]["atomic_number"]
symbol2 = acetic_acid_graph.nodes[node2]["atomic_number"]
bond_order = attrs["bond_order"]
print(
f" Edge {i}: (z={symbol1},id=({node1})) - "
f"(z={symbol2},id=({node2})), bond_order={bond_order}"
)
Using NetworkX for Molecular Analysis (WHY IS THIS HERE?)¶
The following creates a more interesting molecule - benzene - to demonstrate graph analysis:
[ ]:
# Create benzene
benzene_mol = Chem.MolFromSmiles("c1ccccc1")
benzene_mol = Chem.AddHs(benzene_mol)
print("Benzene molecule:")
display(benzene_mol)
# Convert to graph
benzene_graph = molify.rdkit2networkx(benzene_mol)
print(
f"\nNodes: {benzene_graph.number_of_nodes()}, "
f"Edges: {benzene_graph.number_of_edges()}"
)
[10]:
# Find all cycles in benzene
cycles = nx.cycle_basis(benzene_graph)
print(f"Number of cycles found: {len(cycles)}\n")
# The aromatic ring should be the largest cycle
aromatic_ring = max(cycles, key=len)
print(f"Aromatic ring (6-membered): {aromatic_ring}")
# Check bond orders in the aromatic ring
print("\nBond orders in the aromatic ring:")
for i in range(len(aromatic_ring)):
node1 = aromatic_ring[i]
node2 = aromatic_ring[(i + 1) % len(aromatic_ring)]
if benzene_graph.has_edge(node1, node2):
bond_order = benzene_graph[node1][node2]["bond_order"]
print(f" C{node1} - C{node2}: {bond_order} (aromatic)")
Number of cycles found: 1
Aromatic ring (6-membered): [4, 3, 2, 1, 0, 5]
Bond orders in the aromatic ring:
C4 - C3: 1.5 (aromatic)
C3 - C2: 1.5 (aromatic)
C2 - C1: 1.5 (aromatic)
C1 - C0: 1.5 (aromatic)
C0 - C5: 1.5 (aromatic)
C5 - C4: 1.5 (aromatic)
Working with Complex Molecules¶
The following examines a more complex molecule with different bond types - aspirin:
[11]:
# Create aspirin
aspirin_smiles = "CC(=O)Oc1ccccc1C(=O)O"
aspirin_mol = Chem.MolFromSmiles(aspirin_smiles)
aspirin_mol = Chem.AddHs(aspirin_mol)
print("Aspirin molecule:")
display(aspirin_mol)
# Convert to ASE
aspirin_atoms = molify.rdkit2ase(aspirin_mol)
print(f"\n{aspirin_atoms}")
Aspirin molecule:
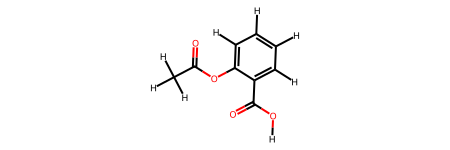
Atoms(symbols='C2O2C7O2H8', pbc=False)
[12]:
# Analyze bond types in aspirin
from collections import Counter
connectivity = aspirin_atoms.info["connectivity"]
bond_orders = [bond_order for _, _, bond_order in connectivity]
bond_counts = Counter(bond_orders)
print("Bond type distribution in aspirin:")
for bond_order, count in sorted(bond_counts.items()):
bond_type = {1.0: "single", 1.5: "aromatic", 2.0: "double", 3.0: "triple"}.get(
bond_order, "unknown"
)
print(f" {bond_type} bonds (order={bond_order}): {count}")
Bond type distribution in aspirin:
single bonds (order=1.0): 13
aromatic bonds (order=1.5): 6
double bonds (order=2.0): 2
Converting Back: ASE/NetworkX → RDKit¶
When we have explicit connectivity information (from RDKit originally), converting back is straightforward:
[13]:
# ASE → RDKit (round-trip)
aspirin_roundtrip = molify.ase2rdkit(aspirin_atoms)
print("Original molecule:")
display(aspirin_mol)
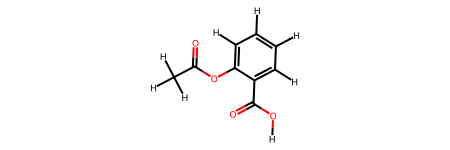
print("\nRound-trip molecule (ASE → RDKit):")
display(aspirin_roundtrip)
Original molecule:
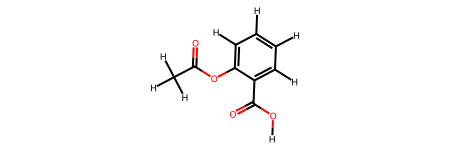
Round-trip molecule (ASE → RDKit):
Key Takeaways¶
What We Learned¶
RDKit has explicit bond information
ASE stores connectivity in
atoms.info['connectivity']as:[(atom_idx_1, atom_idx_2, bond_order), ...]
NetworkX graphs store:
Nodes:
atomic_number,original_index,chargeEdges:
bond_order(always present from RDKit)
Conversions from RDKit are lossless - all chemical information is preserved