NetworkX Interface¶
NetworkX provides a graph-based representation perfect for analyzing molecular topology and connectivity patterns.
For more information about NetworkX, visit https://networkx.org/
[1]:
import ase.build
import networkx as nx
from IPython.display import display
from rdkit.Chem import Draw
import molify
NetworkX → ASE:¶
The networkx graph contains the positional and chemical infromation and can be easily converted to an ASE Atoms object:
[ ]:
# Create a simple molecule and convert to graph
water = molify.smiles2atoms("O")
water_graph = molify.ase2networkx(water)
print(
f"Graph: {water_graph.number_of_nodes()} nodes, "
f"{water_graph.number_of_edges()} edges"
)
# Convert back to ASE
water_back = molify.networkx2ase(water_graph)
print(f"\nBack to ASE: {water_back}")
print(f"Connectivity preserved: {'connectivity' in water_back.info}")
print(f"Number of bonds: {len(water_back.info['connectivity'])}")
NetworkX for Molecular Analysis¶
Graph Algorithms: Finding Cycles¶
[3]:
# Create serotonin (has cycles)
serotonin = molify.smiles2atoms("C1=CC2=C(C=C1O)C(=CN2)CCN")
serotonin_mol = molify.ase2rdkit(serotonin)
display(serotonin_mol)
# Analyze with NetworkX
graph = molify.ase2networkx(serotonin)
print(f"\nSerotonin: {graph.number_of_nodes()} atoms, {graph.number_of_edges()} bonds")
molify.draw_molecular_graph(graph)
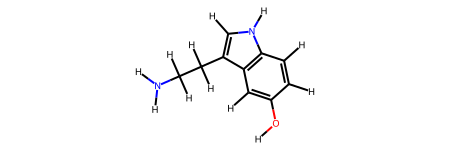
Serotonin: 25 atoms, 26 bonds
[3]:

[4]:
# Find all cycles
cycles = nx.cycle_basis(graph)
print(f"Found {len(cycles)} cycles:\n")
for idx, cycle in enumerate(cycles):
print(f"Cycle {idx + 1} ({len(cycle)} atoms): {cycle}")
# Visualize each cycle
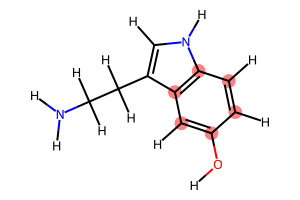
display(Draw.MolToImage(serotonin_mol, highlightAtoms=cycle, size=(300, 200)))
Found 2 cycles:
Cycle 1 (5 atoms): [3, 2, 9, 8, 7]
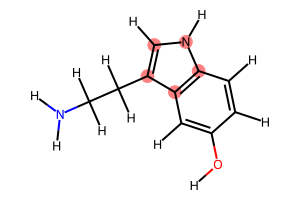
Cycle 2 (6 atoms): [3, 4, 5, 0, 1, 2]
Analyzing Multi-Molecule Systems¶
[ ]:
# Create a water/ethanol mixture
water = molify.smiles2conformers("O", numConfs=10)
ethanol = molify.smiles2conformers("CCO", numConfs=10)
box = molify.pack(data=[water, ethanol], counts=[5, 5], density=800)
# Convert to graph
box_graph = molify.ase2networkx(box)
print(
f"System: {box_graph.number_of_nodes()} atoms, {box_graph.number_of_edges()} bonds"
)
System: 60 atoms, 50 bonds
[ ]:
from collections import Counter
# Find connected components (individual molecules)
components = list(nx.connected_components(box_graph))
print(f"Found {len(components)} separate molecules")
# Analyze molecule sizes
sizes = Counter(len(comp) for comp in components)
print("\nMolecule sizes:")
for size, count in sorted(sizes.items()):
molecule_type = "water (H2O)" if size == 3 else "ethanol (C2H6O)"
print(f" {count} molecules with {size} atoms ({molecule_type})")
NetworkX → RDKit: Guessing Bond Orders from 3D Coordinates¶
Decision Tree: When Does Bond Order Determination Happen?¶
networkx2rdkit(graph, suggestions)
↓
Are all bond_orders present (not None)?
├─ YES → Simple conversion ✅
└─ NO → Bond order determination required ⚠️
↓
Step 1: Try template matching with suggestions
↓
Step 2: Fallback to rdkit.Chem.rdDetermineBonds
(tries charges: 0, ±1, ±2)
Let’s explore each case:
Case 1: Graph Has Explicit Bond Orders¶
When the graph comes from RDKit originally, all bond orders are known:
[7]:
# Create molecule with known bonds
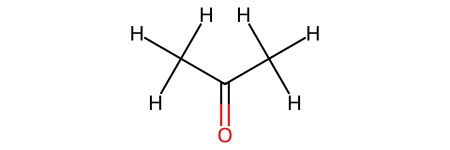
acetone = molify.smiles2atoms("CC(=O)C") # Has a C=O double bond
acetone_graph = molify.ase2networkx(acetone)
# Check bond orders
print("Bond orders in graph:")
for u, v, data in list(acetone_graph.edges(data=True))[:5]:
print(f" {u}-{v}: {data['bond_order']}")
# Convert to RDKit
acetone_mol = molify.networkx2rdkit(acetone_graph)
print("\nConversion successful:")
display(acetone_mol)
Bond orders in graph:
0-1: 1.0
0-4: 1.0
0-5: 1.0
0-6: 1.0
1-2: 2.0
Conversion successful:
Case 2: Graph Has bond_order=None¶
[8]:
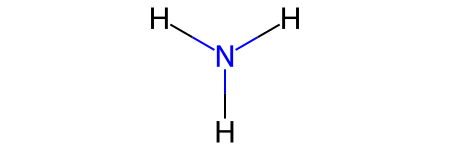
# Create ammonia from ASE (no connectivity info)
ammonia = ase.build.molecule("NH3")
print(f"Ammonia: {ammonia}")
print(f"Has connectivity: {'connectivity' in ammonia.info}")
# Convert to NetworkX - will have bond_order=None
ammonia_graph = molify.ase2networkx(ammonia)
print("\nGraph edge attributes:")
for u, v, data in ammonia_graph.edges(data=True):
print(f" N({u})-H({v}): bond_order = {data['bond_order']}")
Ammonia: Atoms(symbols='NH3', pbc=False)
Has connectivity: False
Graph edge attributes:
N(0)-H(1): bond_order = None
N(0)-H(2): bond_order = None
N(0)-H(3): bond_order = None
Converthing to rdkit with networkx2rdkit will run the bond order determination algorithm.
[9]:
# Convert to RDKit - triggers automatic bond order determination
ammonia_mol = molify.networkx2rdkit(ammonia_graph, suggestions=[])
display(ammonia_mol)
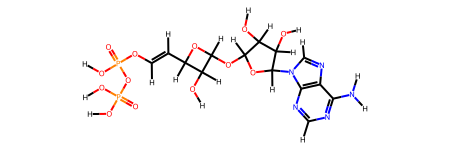
[10]:
SMILES = "OP(=O)(O)OP(=O)(O)OC=CC1C(O)C(OC2C(C(O)C(N3C=NC4=C3N=CN=C4N)O2)O)O1"
molecule = molify.smiles2atoms(SMILES)
molecule.info.pop("connectivity", None) # Remove connectivity info
graph = molify.ase2networkx(molecule)
# the automatic bond order determination can fail for complex molecules
try:
mol = molify.networkx2rdkit(graph, suggestions=[])
except ValueError as e:
print(f"Conversion failed: {e}")
# but if we know the molecule, we can provide it as a suggestion
# You can also provide a list of suggestions for multiple molecules or multiple guesses
mol = molify.networkx2rdkit(graph, suggestions=[SMILES])
display(mol)
# For very large molecules, this can take a while!
Conversion failed: Failed to determine bonds for sub-structure up to charge -2.0 and ['O', 'P', 'O', 'O', 'O', 'P', 'O', 'O', 'O', 'C', 'C', 'C', 'C', 'O', 'C', 'O', 'C', 'C', 'C', 'O', 'C', 'N', 'C', 'N', 'C', 'C', 'N', 'C', 'N', 'C', 'N', 'O', 'O', 'O', 'H', 'H', 'H', 'H', 'H', 'H', 'H', 'H', 'H', 'H', 'H', 'H', 'H', 'H', 'H', 'H', 'H', 'H', 'H']
Key Takeaways¶
What We Learned¶
NetworkX → ASE: Simple data transfer
NetworkX graphs are great for:
Finding cycles (
nx.cycle_basis)Counting molecules (
nx.connected_components)Analyzing topology
NetworkX → RDKit complexity depends on
bond_order:If all present → direct conversion
If any
None→ Bond order determination
Bond order determination uses two steps:
Step 1: Template matching with
suggestions(SMILES list)Step 2: Fallback to
rdkit.Chem.rdDetermineBondsTries charges: 0, ±1, ±2
Special handling for ions
The “suggestions” parameter:
List of SMILES strings
Can be complete molecule or substructures
Helps ensure chemical correctness