RDKit Interface¶
RDKit is a powerful cheminformatics toolkit that provides extensive functionality for molecular manipulation, analysis, and property calculation. For more information about RDKit, please visit https://www.rdkit.org/docs/index.html
[1]:
from ase.collections import g2
from IPython.display import display
import rdkit2ase
rdkit2ase uses RDKit to create ase.Atoms objects from SMILES strings and convert them to ase.Atoms objects. But you can also use rdkit2ase to convert ase.Atoms objects back to RDKit Mol objects.
[2]:
nitroanilin = rdkit2ase.smiles2atoms(smiles="c1cc(c(cc1)N)N(=O)=O")
mol = rdkit2ase.ase2rdkit(nitroanilin)
print(mol)
mol
<rdkit.Chem.rdchem.Mol object at 0x11c2f23b0>
[2]:
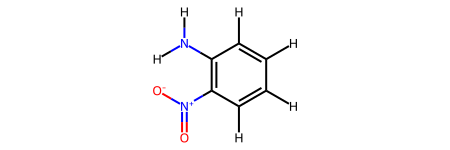
For example, you can convert ase.Atoms objects from the ase.collections.g2 database to RDKit Mol objects and display them or use any other RDKit functionality:
[3]:
for idx, (atoms, name) in enumerate(zip(g2, g2.names)):
print(name)
display(rdkit2ase.ase2rdkit(atoms, suggestions=[]))
if idx == 10:
break
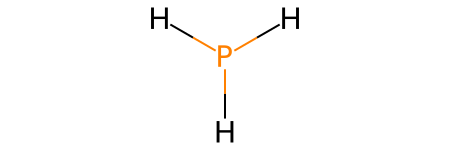
PH3
P2

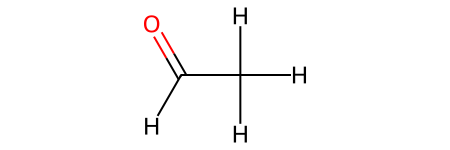
CH3CHO
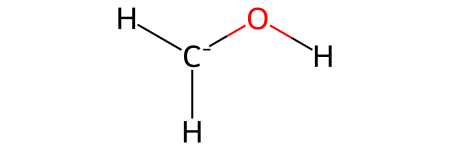
H2COH
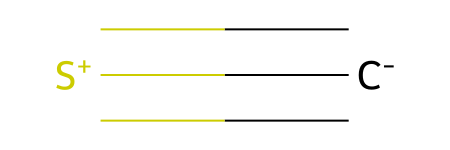
CS
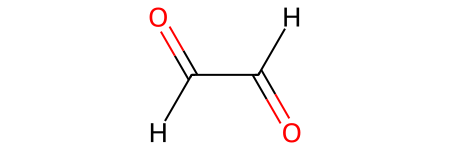
OCHCHO
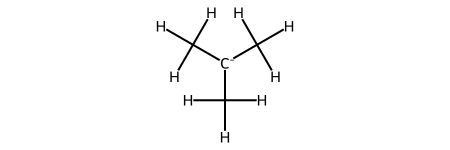
C3H9C
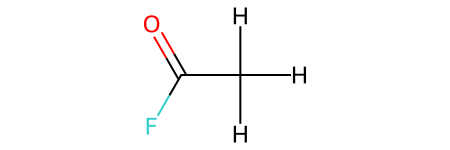
CH3COF
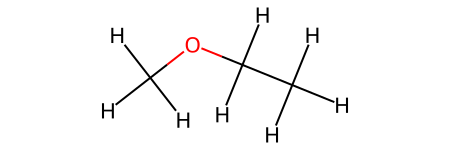
CH3CH2OCH3
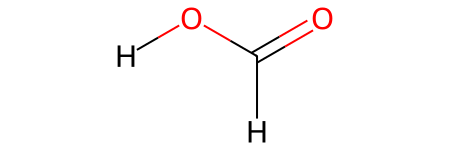
HCOOH
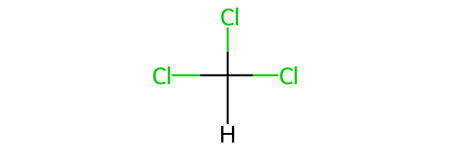
HCCl3
[ ]: